I believe that autism has been ‘discovered’ in the wrong order. Autistic thought, loosely labeled as the broader autism phenotype (BAP), is a perfectly normal and reasonable example of neurodiversity, within the normal range of human thought. I see autistic disabilities as being driven by sensory integration issues to which those who fall within the BAP are more vulnerable. In this paradigm it is reasonable to expect the medical and psychiatric professions to ‘discover’ ASD in reverse order of severity, i.e. defining first the more severe disabilities, then lesser disabling cases, and then the population out of which these cases arise. It is also reasonable that the disabling ‘comorbidities’ of the BAP are therefore not essential features of this neurology, but rather, that they may be medical conditions worthy of amelioration, and that ameliorating these conditions would not change the underlying BAP way of thinking, but instead would allow it to flourish.
Minicolumns and The Brain
The neocortex (also known as the isocortex) is the 2 to 4mm tick top layer of the cerebral hemispheres and outer layer of the cerebral cortex. This layer is responsible for higher functions such as sensory perception, generation of motor commands, spatial reasoning, conscious thought, and language. The neocortex itself is divided into six layers, although there are no borders between these layers, and neuron dendrites and axons cross multiple layers.
The basic anatomical and physiological unit of the neocortex is the minicolumn. (Casanova 2006) As its name suggests, a minicolumn is a vertically aligned collection of cells and their projected connections. It is the smallest unit capable of information processing in the brain - resembling a mini-processor in function - receiving stimuli from elsewhere, processing information, and providing the capability of a response. Information is transmitted through the core of the minicolumn and is prevented from activating neighboring minicolumns by surrounding inhibitory fibers (interneuronal projections).
Pyramidal cells make up approximately 80% of the neurons of the cortex, and are the heart of the information processing capability of the minicolumn. Pyramidal cell somata (cell bodies) in layers III, V and VI are vertically oriented. Their dendrites and axon trees cross at least three layers, and in many cases all of the layers of the neocortex. They release glutamate as their neurotransmitter, and are the major excitatory component of the cortex.
Another component of minicolumns are various types of GABAergic inhibitory interneurons (i.e. neurons that produce GABA as their neurotransmitter), which tend to be aligned one on top of the other and modulate the activity of minicolumn pyramidal cells. Double bouquet cells are present in all layers, but are most dense in layers II and III. The axon bundles (long projections that conduct electrical impulses away from the cell body) of these cells are projected deep into the cortex from layer II to layer V, terminating on pyramidal cells as well as other inhibitory interneurons. They create a narrow vertical stream of inhibition through the cortex, as well as a vertically directed disinhibition of those pyramidal cells upon which the other inhibitory interneurons project. Other interneurons include Chandelier cells, which synapse directly onto the axon hillock (base of the axon projection from the cell body) of pyramidal cells, modulating cell output and participating in intra-columnar inhibition, and basket cells, which contacts the soma and dendrites of pyramidal cells, modulating input to these cells. Although these interneurons make up a small fraction of the total number of minicolumn cells, they play a significant role in finely tuning cortical information processing. A simplified representation of a minicolumn is thus of a processing core surrounded by a GABAergic interneuron circumferential zone of inhibitory and disinhibitory activity.

(Figure 2 Source – TMS Research Proposal in preparation for submission, pg 2, with permission)
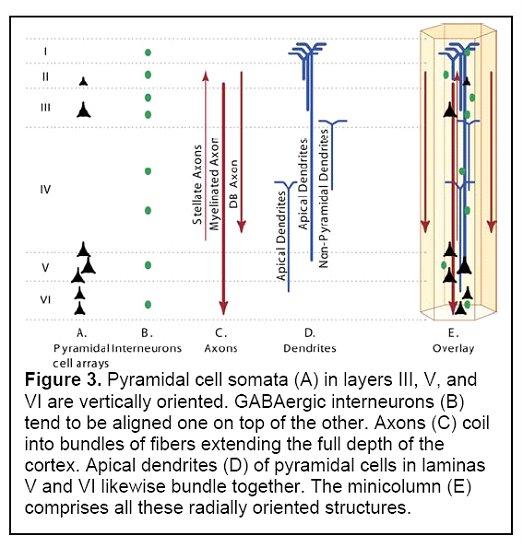
(Figure 3 Source – TMS Research proposal in preparation for submission, pg 3, with permission)
Minicolumns are the basic organizational unit of the cortex. It is an increase in the number of minicolumns, expanding the area covered by the isocortex, that is responsible for the historical process of encephalization, i.e. the increase in size of the brain to a degree greater than that expected based on body size (in modern humans, brain size is three to five times greater than expected when comparing to mammals of equal body mass) (Casanova - Big Brains Manuscript, in preparation for submission). A variable number of minicolumns are dynamically clustered with neighbouring minicolumns into macrocolumns, and from there in networks of macrocolumns and regions of functional differentiation within the brain. The minicolumn-macrocolumn relationship may be linked in part to both the termination of projections from the thalamus, which span a fixed distance and may serve to link together minicolumns that receive input from the same thalamocortical fibers, and by the effects of serotonin, changing columnar development in the cortex during brain development (Casanova 2006).
This minicolumn organizational structure conveys certain advantages. Through limiting connectivity within the brain (connectivity requires a significant amount of energy), the result is a reduction in metabolic expenditure. Instead of connecting every cell within the cortex to different brain regions, projections are organized into modules. Single cells project to target sites, and information gained or transmitted is transferred to other neurons within the same modules. Another advantage is plasticity, or the ability of the brain to naturally reorganize connections. Similarities between minicolumns suggest that they are capable of performing similar transformations on incoming information, allowing them to replace each other in case of injury or to adapt to changing requirements. This is not to suggest that all minicolumns are identical. There are variations within different regions, but these may arise from variations in input, output targets, interconnectivity, and inhibition (Casanova - Big Brains Manuscript, in preparation for submission).
So, what does this have to do with autism?
Analysis has determined that minicolumns in the brains of autistic individuals tend to be smaller in size, although with the same total number of cells per column (Casanova et al, 2002a and Casanova et al 2002b). Given that autistic brains tend to be larger than average, the results indicate that autistics also have a higher number of minicolumns. In addition, the neurons within these individual minicolumns tend to be reduced in size.
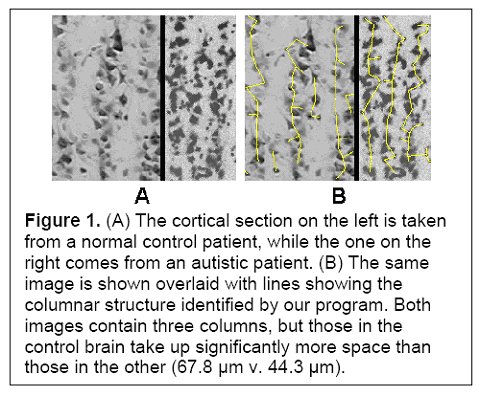
(Figure 1 Source – TMS Research proposal in preparation for submission, pg 2, with permission)
Smaller minicolumns would skew information processing (noise/signal) in favour of signal, potentially enhancing the ability to process stimuli that require discrimination, but also potentially at the expense of generalizing the salience of a particular stimulus. Smaller and more densely packed minicolumns could also allow for more complex information processing. As an example, the smaller minicolumns in the visual cortex may support added functionality, e.g. depth or color perception. This may be due to the overlap of neuronal projections allowed for when neuronal dendrites and axons remain the same size but the distance to neighbouring minicolumns is reduced (Casanova - Abnormalities Of Cortical Circuitry In The Brains Of Autistic Individuals).
These enhancements come at a cost. In autism, minicolumn size reductions result primarily from reductions in the peripheral zone of inhibitory and disinhibitory activity. The inhibitory fibers act to keep the stimuli within individual minicolumns, and the reduction in this space results in stimuli no longer being contained. Instead, they can overflow to adjacent minicolumns, providing an amplifier effect, which may explain hypersensitivity in some autistics. Thalamic input may also be amplified if thalamic terminal fields remain the same size and therefore result in more minicolumns per stimulated macrocolumn. Each minicolumn’s response to thalamic input is also modulated by the activity of neighbouring columns to a greater or lesser degree, allowing for gradations of response, so a reduction in GABAergic inhibitory activity could also result in a loss of inhibition and greater amplification (see Figure 1). Stimuli ‘spill’ and amplification could result in increased incidence of seizures in autistics.

(Casanova 2006, pg 3, with permission)
An additional cost is that imposed by a reduction in neuron size. The ability of a neuron to sustain a connection over distance is related to the size of its cell body. Smaller neurons result in a metabolic bias favouring shorter connections at the expense of both longer distance and inter-hemispheral connectivity. The result is that autistic brains have a bias towards local (intra-regional) over global (inter-regional) connectivity and processing. Short intra-regional processing functions include mathematical calculations and visual processing. Cognitive functions that require inter-regional processing would be less metabolically efficient, including language, face recognition, and joint attention (Casanova - Abnormalities Of Cortical Circuitry In The Brains Of Autistic Individuals).
The total number of minicolumns in the human brain is defined during the first forty days of fetal development. Symmetrical divisions of germinal cells determine the total number of minicolumns, and a second phase of asymmetrical divisions provides for cell migration into the cortex, with successive divisions determining the number of cells in each minicolumn. This is a complex process, defined by a large number of genes interacting with the environment. The result is that a higher number of minicolumns is not something that someone can ‘acquire’ or be vulnerable to within the post-natal period. It is possible for disruptions during the narrow window of fetal development (e.g. rubella, thalidomide, tuberous sclerosis) to also result in an increase in the number of minicolumns, by influencing this process (Casanova - Abnormalities Of Cortical Circuitry In The Brains Of Autistic Individuals; Casanova 2006).
One point that is important to make is that while narrower minicolumns is a factor in autism, evidence suggests that it is not related to mental retardation. In Down Syndrome, as an example, minicolumn width is normal, despite the smaller brain sizes of Down Syndrome patients.
Implications
Reduced minicolumn width appears to be a prerequisite for autism. But, the reported minicolumn widths found within autistic brains are still within the normal distribution of minicolumnar width, albeit at the tail end (Casanova 2006). This suggests to me that the existence of narrow minicolumns is not enough by itself to result in an ASD diagnosis. The key instead appears to be a reduction in inhibition within minicolumns, rather than width alone. Reduced width increases the consequences of reduced inhibition, but does not automatically cause it. In effect, a brain with narrower minicolumns may be less robust, and therefore more vulnerable to the complications that could come with deviation from the narrow tolerances within which the brain functions. In a brain with wider minicolumns, a loss of inhibition would not have as significant an impact, as minicolumnar width (and therefore distance between minicolumn information processing cores) would still exist to reduce intercolumnal spill, and thalamic projections would result in fewer minicolumns per macrocolumn to be affected.
It is a logical assumption that minicolumn development in MZ twins would be similar (same alleles, very minimal epigenetic differences during the early weeks of fetal development). But MZ twins do not always share the same ASD diagnosis. In cases in which one twin has an ASD diagnosis, the chance of the second twin also having an ASD diagnosis (not necessarily of the same severity) is 60%, and the chance of the second twin being within the broader autism phenotype is 92%. While ASD clearly has an inheritable component (the prevalence of ASD in the population is 0.6%, but within families with an ASD child, the chance of subsequent children being ASD is up to 10%), genetics alone is not sufficient to explain a diagnosis in all cases. This, combined with the fact that reduced autism minicolumn widths still fall within the normal range suggests to me that the issue in autism relates to loss of inhibition, to which autistics are more vulnerable than average, i.e. reduced minicolumnar width is a precondition for autism but a 'second hit' is still required. If that second hit were purely genetic, as distinct from epigenetic or environmental, then presumably both MZ twins should be similarly affected. This is clearly not the case a significant percentage of the time.
Support for this may come from studies of the brains of MZ twins discordant for autism. In Kates et al 2003, a comparison of MZ twins discordant for autism indicated that both the autistic and non-autistic (broader autism phenotype) twins showed no significant differences in cerebral gray matter. This suggests a structural similarity in MZ twins, and the possibility of a requirement for a ‘second hit’ to explain the discordant outcomes.
We can speculate on what that ‘second hit’ may be, and indeed the speculation as to a) whether a second hit exists and b) the causes, if any, are among the most controversial subjects of debate within the community of those linked by autism. I will look a bit more closely in this direction in a post in the near future.
What is clear from above though is that if narrow minicolumnar width is a) a precondition for ASD and b) not sufficient by itself to cause ASD, then this should have an impact on ASD research. If narrow minicolumns creates a vulnerability that wider minicolumns do not, then demonstrating lack of effect in those who are invulnerable does not demonstrate lack of effect in those who may be vulnerable. In many cases the correct population in causation analyses (e.g. epidemiological studies?) would not be the wider population, but instead the sub-section of the population who are actually vulnerable. And in looking for causes of autistic disability, we may want to look more closely at those events and/or stimuli that may reduce inhibition within the brain and/or further discourage global connectivity.
It is also a reasonable proposition is that a ‘cure’ for autism would not alter the underlying narrow minicolumnar structure of the brain. Instead, it would allow this structure to function more effectively, maintaining its strengths while reducing or eliminating the more disabling features of ASD. A cure would not change who one is, which is one of the fears of the neurodiversity community. Instead, it would allow autistic thought to function without the disabling comorbidities, serving to maintain neurodiversity and allowing it to flourish.
Update - Related post:
Autism and the Evolution of the Brain (Oct 13, 2006)

26 comments:
Yes, at first glance, Casanova's proposed treatment looks as if it could be helpful for people who have major hypersensitivity issues, but I do wonder if it might turn out to have unintended consequences. Have you read The Poisonwood Bible? One of the characters, Adah, is apparently a hyperlexic Aspie (though the author doesn't use psychological terms to describe her). Adah walks very awkwardly. An experimental treatment greatly improves her gait, but she discovers afterward that she has lost (perhaps forever) the beauty and fascination that she had found in words.
Hi Bonnie,
Thanks for stopping by and for the comment. I haven't read The Poisonwood Bible, but I have also wondered about the possibility that treatment (not discussed in this post) may have unintended consequences related to some of the more beneficial autistic traits. One of the results that I would hope for from research and testing is an answer to this question.
I would speculate though that these unintended consequences may not materialize. The underlying narrow minicolumn structure, the different macrocolumnar arrangements and different thalamocortical links - i.e. the presumed foundations of the neurodiversity of thought in those with ASD - would continue to exist post-treatment. What I would anticipate is that with enhanced inhibitory functioning that ‘accidental spill’ would diminish. But all of the other increased local connectivity would presumably continue to exist. I’m not sure that ‘accidental spill’ would necessarily contribute to richness of thought, as by its nature it would be random and extremely fleeting.
What I would suggest though is that this should be a question for research to answer rather than a reason not to proceed with research (and I am definitely not trying to imply that you are suggesting anything above other than a perfectly valid cautionary note, with which I would agree).
I like your article, I really love the part about minicolumn number being set by 40 days. But you are wrong that a second hit would be needed to explain the difference between monozygotic twins. Just as a "second hit" is not needed to explain the difference in fingerprints between MZ twins. Some of the construction of the brain is very random. It just happens a particular way, in the same way that fingerprints happen. They are not 100% genetically determined, obviously, or twins would have identical fingerprints.
Also, there can be significant differences in the womb, so I heard one time over at the MIND Institute. The position of one baby as compared to it's twin can affect the amount of nutrition it gets from the placenta.
If one twin kicks more than the other, it's brain will be wired differently, in subtle ways, of course.
Development is ultra complicated, not just a matter of a set of genetic blueprints that are followed, not by a long shot.
Which is not to say that no research should be done into seeing if drug or chemical exposure can increase the likelihood of an autism diagnosis. The odds of that thing being anything found in a vaccine are nil. The odds of it being something outside of a vaccine are massive.
The odds that chelation or GFCF diet or B12 shots truly have an impact on an autistic brain....making it more normal? Gimme a stinkin' break. This is a massive con job... but then I'm off on a rant...
Changes that happen early in development are massively more important than changes that happen later because of the "downstream effects". You can't undo a change that happened at 6 weeks into gestation, after the kid is born. You can find ways to mitigate the existing condition, or to aggravate it, but you can't undo the foundation. This is why moms shouldn't drink alcohol while pregnant, it's not a matter of sobering up the baby ("chelating" the alcohol out of him), the damage is done, the shape of the kids brain is mostly set.
That's what totally ticks me off about these... these...freaks.. saying they should chelate a kid whose mom got Rhogam shots. OK, end rant. Thanks for alerting me to your blog entry.
I don't think they should aim for a cure for autism until they understand what they are curing.
Hi Camille,
Thanks for stopping by and for the comment (and for holding back ;-) ). We’ll have to disagree on the MZ twin issue, but I have no issue there.
My understanding regarding genetics is that events are ‘programmed’, but are not under total control. There is simply not enough DNA to control the exact placement and exact structure of every cell or every sub-cellular structure in the body. As such, to cover fingerprints first, genes determine basic fingerprint structure and style (e.g. arch vs. tented arch vs. whorl) but exact fingerprints still have an element of randomness within them.
I’m under the understanding that brain structure is a similar case. Individual minicolumns could presumably have a slight randomness in placement, but the randomness would cover matters like having 600,000,068 vs 600,000,341 minicolumns, not narrow minicolumns vs. minicolumns of average width. Within each minicolumn, presumably each cell of the same type would be governed by the same strands of genetic material, subject to any changes that took place during cell division and any subsequent epigenetic and environmental impacts. There is not one strand of genetic material governing pyramidal cell 24,567,409 and a different strand governing pyramidal cell 153,768,093 of the same type (okay there are different strands, because they are physically located within different cells, but it is the same alleles in both cases).
If the minicolumn explanation is correct, then narrow minicolumns is a precondition for ASD. But ASD would presumably not be caused by slight random variations in minicolumn placement, especially if the minicolumns are similar in size and structure. Instead, the variation would come from significant performance changes within minicolumns and within the connections (or lack thereof) of thousands or even millions of columns, which would require a cause that could simultaneously impact a very large number of cells (albeit not necessarily over a large expanse of brain geography). Minor microscopic variations in columnar placement would not be material, in the same way that we would presumably not be affected if a particular column ceased to function. The variation would have to be significant in scope and scale (although presumably microscopic in size).
I do agree that issues such as nutrition can affect developing twins, and that the effects may not be subtle. I also agree that intervention will not make an autistic brain more ‘normal’, if by normal you mean altering the brain into a more average minicolumnar configuration. But if the science is correct, then this ASD configuration is already within the ‘normal’ range (albeit at the tail end of the normal distribution curve) and is a pre-condition for ASD, but is not enough to cause ASD. The issue is not structure but a reduction in inhibition. You can have this minicolumnar configuration and not qualify for an ASD diagnosis, presumably because there is no failure in inhibition and connectivity. As such, the changes that cause ASD must be at a cellular level or at a level that impacts inhibition and connectivity, subject to the precondition of having this narrow minicolumn structure.
My understanding is that connectivity is a dynamic and ongoing process, albeit building on what has gone before. As such, in the case of MZ twins, the genes that govern both the individual cells and the mechanisms that underlie the processes of connectivity would be the same. The difference would be due to ongoing epigenetic or environmental factors within the brain (plus variations in existing connectivity driven by 'experience'), and they would presumably have to be widespread and exist over time. This to me would be the ‘second hit’. And I would suggest that the effects of this 'second hit', may be at least partially reversible or ameliorable, but would not change the underlying narrow minicolumnar structure.
Ian,
This is a fantastic post. Camille beat me to the fingerprint point, and obviously I think the brain development to be far more complex. The only issue I have with all the discussion is what the definition of "environmental factors" as a second influence could be. I see it as nearly infinite in possibility especially within the first 40 days: nutrition, random variation, temperature, pH, blood pressure, movement (maybe even gravity), pathogens, sound, hormones, etc. You get the idea. I think a "second hit" could be incredibly nebulous, although it's undoubtedly there in some form (as evidenced by fingerprints in MZ twins). I wonder what Cassanova's thoughts are on this aspect.
Hi All,
Okay, this is beginning to feel like a scene the Monty Python film “The Meaning of Life” – the one in the boardroom where the participants were discussing the meaning of life. One of them gave an absolutely brilliant explanation of the meaning of life (I highly recommend the movie for this explanation alone), followed by the statement “And people aren’t wearing enough hats.” Of course the rest immediately launch into a discussion about hats, ignoring the meaning of life (as predicted in the explanation provided). I introduced the hats, i.e. MZ twins, and everyone immediately leapt there. Mea culpa. I’ll now follow, and then come back to the central point of the post.
Joseph wrote:
"Isn’t there research that does document differences in grey and white matter volume in discordant MZ twins?" The Kates et al link above found no significant differences in gray matter in discordant MZ twins. Minicolumns are gray matter. White matter is a separate issue, driven by and related to connectivity (more below).
"I bet concordance of minicolumn size is not 100% in MZ twins." Possibly, but not too many of them are volunteering for simultaneous autopsies to find out (sorry, it’s late and I’m being flippant). Regardless, the point is not that minicolumn size drives ASD, but that minicolumn size is a prerequisite but not sufficient by itself. A smaller average minicolumn width in the non-ASD MZ twin does not invalidate this point, and minicolumn size is not related to 'severity'.
"This doesn't even consider random factors after birth. For example, why is the concordance of intelligence in MZ twins reared together 80% whereas the concordance for those reared apart is 70%? A second hit?" I would suggest that the difference in this case is experientially caused. If autism is experientially caused, as distinct from ASD brain development being impacted by ‘experience’ (suggested in Kates et al to explain cerebellar differences between discordant MZ twins), I would be very interested in finding out more.
Human growth hormone is not totally predictive because it is one factor among several that affect the ultimate height that a person may attain. Minicolumns are more predictive in that – as per your analogy – they are part of the height. But reduced minicolumn width is a necessary prerequisite for ASD and a factor driving neurodiversity, but is not sufficient by itself to cause ASD. This is one of the central points of the post.
Reduced blood flow would be affected by both ‘supply’ and ‘demand’. From a supply perspective, blood flow would either be adequate or result in brain damage (lack of oxygen and nutrition). From a demand perspective, developing cell requirements would help drive blood supply development, so underdevelopment in a region of the brain would reduce demand and therefore supply. To my knowledge, no one has demonstrated that autism results from lack of blood supply to the brain in any reasonable percentage of cases, and if it did then cell damage should be both apparent and detectable.
"Anyway, what Casanova is doing is basically figuring out the underpinnings of autistic neurodiversity, which is a concept that came about from autistic people's intuition." Agreed, at least regarding the underpinning of neurodiversity. This is another of the points of the post.
Do’C also wrote about the ‘second hit’ and brought up fingerprints. We’re probably all in agreement that brain development is far more complex. Having said that, while we can speculate on whether the second hit takes place within the 40 day window or not, let’s look for a moment at the requirements for a second hit to be material rather than inconsequential. One of the rationales for minicolumn existence is redundancy. So for a second hit to be material it must affect not just one or a small number of minicolumns, but must be significant in impact, and in effect must ‘permanently’ override normal development.
Both ASD and non-ASD minicolumns have the same number of cells, and the same vertical interneuronal distances (Casanova 2006). The difference between ASD and non-ASD is presumed to be both reduced minicolumn width (a prerequisite, but not sufficient in and of itself) and reduced inhibition (required). Reduced inhibition would presumably be either a chemical process (e.g. a reduction in GABAergic inhibitory activity) and/or cellular/physical (e.g. a reduced number or expanse of dendrites, other cell structure, etc.). I’m sure I’m missing some other options here.
Regardless, the ‘cause’ and its impact would have to be significant in scope and be sustained to make a material difference across a significant number of minicolumns, and it would have to differ within MZ twins who share the same genetic and epigenetic heritage (environment may differ) in utero, and would presumably not significantly differ from an epigenetic perspective within the first few years of life. Are there isolated cases of significant epigenetic differences in utero or during the first two years? Undoubtedly. But at a level of 40% (ASD vs non-ASD, even if ignoring variations in diagnosis within the concordant 60%)? Unlikely, given research indicating only minor epigenetic differences within the early years of life. An isolated aberration isn’t enough in a dynamic system with built-in redundancy and plasticity to explain the percentage of cases of discordant diagnosis. The difference has to be greater in scope and impact than that implied by similar but not exact fingerprint patterns in MZ twins.
Meanwhile, the point of introducing MZ twins into the equation was to demonstrate that genes by themselves are not enough to account for all cases of ASD, and to point out the Kates research that indicates no significant differences in cerebral gray matter in discordant MZ twins. Beyond that, twins are irrelevant to the case being made, that a) ASD has a minicolumnal underpinning, b) this underpinning is required (i.e. no narrow minicolumns means no ASD), c) it originates in the first 40 days of fetal development (i.e. it is not itself acquired post-natally), d) that this difference falls within the normal range (i.e. that having it does not ‘cause’ a diagnosis, although it may very well result in diversity of thought and cognition, i.e. neurodiversity), e) that something else is therefore required (with no significant speculation by me yet as to what that something else may be, other than to generically label it as a ‘second hit’), and f) that research needs to prove or exclude causality among the population of the vulnerable, i.e. proving that something does not cause ASD in those who are invulnerable does not prove that it does not cause ASD in those who are vulnerable.
Now, getting back to that hat question… ;-)
for a second hit to be material it must affect not just one or a small number of minicolumns, but must be significant in impact, and in effect must ‘permanently’ override normal development.
You're making an unsupported assumption here, Ian, which is that the development of autistic traits is "abnormal" development. How do we know that it isn't the natural expression of the genes involved? Getting back to the twins, it's possible that the non-autistic twin could be the one who takes an environmental "second hit" that makes him turn out not to be autistic. We simply don't know. As Joseph pointed out, Casanova's research is only just beginning to identify the underpinnings of neurodiversity.
It will be interesting to see what happens with the research. I hope that Casanova will be very careful about informed consent and will not experiment on any person who has communication difficulties that could interfere with telling him of negative effects, should they occur.
Hi Bonnie,
I'll agree that from a pure logic standpoint it is an unsupported assumption that the hit must be to make the non-ASD (but still neurodiverse) ASD, rather than the reverse. Logically the 'hit' could be in either direction (A->B or B->A), regardless of whether the person is half of a twin set.
I would suggest though that the assumption is reasonable – invoking Occam’s razor, that the most simple explanation is the most likely – in that it is more likely that a ‘normal’ development path (normal in terms of structure, not outcome) is altered to follow a divergent path (to ASD) than that development first diverges from the norm (to ASD) and is then altered back to the norm. Having said that, as a wise friend commented once in rebuttal to an invocation of Occam’s razor, "Everything should be made as simple as possible, but not simpler. —Albert Einstein’s comment on Occam’s Razor". I recognize that reasonable is in the eye of the beholder and is not the same as 'proven'.
To be clear in the above, I am not suggesting that any and all ASD traits are ‘abnormal’. In contrast, I would expect that a brain structure based on narrower minicolumns would result in different traits that are perfectly normal for that structure and should not be ‘corrected’ or ‘fixed’.
Hi
I disagree respectfully with you, Camille and DoC.
I consider that yours is and opinion based on personal analysis. Mine is different.
When genetics and proteomics is considered- including the metabolic pathways involved - and clinical studies can be designed properly several environmental components arose: heavy metals/xenobiotics and others such as Al, PCBs and derivatives, organic chemicals and infections are the main suspects because of impact in overall immune health in NT population and in CNS, and there are a lot of published research about. Biological plausibility is an important aspect to study, especially if there is anecdotical evidence to consider. Actual field of neuroimmunotoxicology presents a different view the HM, including its effect and importance in epigenetics.
Even more, it has not been enough/properly studied- at the molecular level- yet to have enough clues to say one way or the another in autistic people.
There are some clues on the negatives effects of vaccines on susceptible people, specially related to autoimmune conditions. Some of them
Can unresolved infection precipitate autoimmune disease?.
Severe Reactions Associated With Diphtheria-Tetanus-Pertussis Vaccine: Detailed Study of Children With Seizures, Hypotonic-Hyporesponsive Episodes, High Fevers, and Persistent Crying.
Neurologic Events Following Diphtheria-Tetanus-Pertussis Immunization.
Infection, vaccines and other environmental triggers of autoimmunity.
Autoimmune hazards of hepatitis B vaccine.
In genetically susceptible individuals or together with some other triggers the combination of toxoids/atenuatted viruses plus adjuvants might confer the risk of developing a continuous autoimmune response in an individual.
Autoimmune diseases and vaccinations.
On adjuvants:
-Biomed Pharmacother. 2004 Jun;58(5):325-37.
Autoimmunity induced by adjuvant hydrocarbon oil components of vaccine.
Kuroda Y, Nacionales DC, Akaogi J, Reeves WH, Satoh M.
-Toxicology. 2004 Mar 15;196(3):211-6.
Autoimmunity, environmental exposure and vaccination: is there a link?
Ravel G, Christ M, Horand F, Descotes J.
-J Autoimmun. 2003 Aug;21(1):1-9.
Induction of lupus autoantibodies by adjuvants.
Satoh M, Kuroda Y, Yoshida H, Behney KM, Mizutani A, Akaogi J, Nacionales DC, Lorenson TD, Rosenbauer RJ, Reeves WH.
The importance of BNDF in the development of somatosensory cortex is important and it has been published that is altered in ASD.
If heavy metal/Al transport /excretion systems are blocked or not in proper functioning (not studied yet in ASD at the whole metabolic route, only one paper published on glutathione methyl S transferase 1 on genetics and manuscripts of Dr James need further confirmation, but they are clues), HM are bioaccumulated from the source you mentioned (not studied yet in ASD with a method/approach trustable) if viral/bacterian components (specially related to proteins) can have high impact because of an immune dysfunctional system such as common childhood viral/bacterian issues not targeted by vaccines have (not enough studied yet in ASD), if adjuvants have potential as autoimmune disease triggers (not enough studied yet in ASD); I can´t consider that the probabilities of vaccines as a COLLABORATOR- but not causes for me- are so low as you pointed out. Environmental pollution (in air, food and water) is other source to take into account, perhaps with a big impact because of daily exposure. Considering biological plausability and metabolic targets the main HMs candidates seem to be Hg, Cd, Pb, As, Mn-mainly- and other metals (Al). Considering bacterian infections, viruses, toxoids and strep are enough wide to have an enourmous impact. But the main point for me is the target of all these environmental components, that is the immune system and the intrincate relation between the gut, the immune system and the brain. Therefore, focusing in the research of oxidative stress, immune system, gut and brain- structure and physiology mainly neurotransmitters- in ASD for one side plus all the neurocognitive facts that can be systematized in children/teens and adults, the potential stressors could be better defined and identified, especially with the tools of genetic epidemiology. Therefore I honestly do not see the situation as nebulous. What I see in the mainstreamed published science is a situation with a lot of clues, without an unified “everything theory” to test, or simply no theory at all. It is presented the how/what is different in ASD vs NT but it is not the why- and the attribution to genetics is for me nebulous.
There are several reports about the developmental brain as target of different chemicals and implicancies of transport of HM .
Purkinje cell and cerebellar effects following developmental exposure to PCBs and/or MeHg.
Behavioral changes in metallothionein-null mice after the cessation of long-term low-level exposure to mercury vapor.
Transport of a neurotoxicant by molecular mimicry: the methylmercury-L-cysteine complex is a substrate for human L-type large neutral amino acid transporter (LAT) 1 and LAT2
About treatments to ameliorate the impact of altered physiology, this is a totally different world. However, honestly, the approach for me is not intended as an attemp to render my childs brain more “normal”, but more physiologically optimal in functioning with the actual structure his brain has.
Prenatal exposure is one of the worst impacts of xenobiotics- but not the only one. I agree with you in this topic.
Work of Dr Courchesne
Brain overgrowth in autism during a critical time in development: implications for frontal pyramidal neuron and interneuron development and connectivity.
Brain development in autism: early overgrowth followed by premature arrest of growth.
The autistic brain: birth through adulthood.
and links cited there.
A good recent review on genetics- exclusively-
The Developmental Neurobiology of Autism Spectrum Disorder.
However, environment is cited, and the lack of knowledge of etiologic factors is pointed out several times. From this last one
“There has also been considerable attention to the possible contribution of environmental factors (London, 2000). Several prenatal exposures have been associated empirically with ASD including thalidomide, certain viral infections, and maternal anticonvulsants, especially valproic acid (Trottier et al., 1999; Arndt et al., 2005). Although these factors independently account for few cases, environmental factors may interact with genetic susceptibility to increase the likelihood of ASD. For example, some data implicate a possible role of immune factors, including an increased family history of autoimmune diseases and presence of autoantibodies to neural antigens (Ashwood and Van de Water, 2004 -The immune response in autism: a new frontier for autism research.
J Leukoc Biol. 2006 Jul;80(1):1-15. Epub 2006 May 12.; Connolly et al., 2006.
New Thinking on Neurodevelopment .
Concentrations of environmental chemicals associated with neurodevelopmental effects in U.S. population .
Other manuscript published in the last 5 years
Ment Retard Dev Disabil Res Rev. 2002;8(3):188-97.
Environmental factors associated with a spectrum of neurodevelopmental deficits.Mendola P, Selevan SG, Gutter S, Rice D.
A number of environmental agents have been shown to demonstrate neurotoxic effects either in human or laboratory animal studies. Critical windows of vulnerability to the effects of these agents occur both pre- and postnatally. The nervous system is relatively unique in that different parts are responsible for different functional domains, and these develop at different times (e.g., motor control, sensory, intelligence and attention). In addition, the many cell types in the brain have different windows of vulnerability with varying sensitivities to environmental agents. This review focuses on two environmental agents, lead and methylmercury, to illustrate the neurobehavioral and cognitive effects that can result from early life exposures. Special attention is paid to distinguishing between the effects detected following episodes of poisoning and those detected following lower dose exposures. Perinatal and childhood exposure to high doses of lead results in encephalopathy and convulsions. Lower-dose lead exposures have been associated with impairment in intellectual function and attention. At high levels of prenatal exposure, methylmercury produces mental retardation, cerebral palsy and visual and auditory deficits in children of exposed mothers. At lower levels of methylmercury exposure, the effects in children have been more subtle. Other environmental neurotoxicants that have been shown to produce developmental neurotoxicity include polychlorinated biphenyls (PCBs), dioxins, pesticides, ionizing radiation, environmental tobacco smoke, and maternal use of alcohol, tobacco, marijuana and cocaine. Exposure to environmental agents with neurotoxic effects can result in a spectrum of adverse outcomes from severe mental retardation and disability to more subtle changes in function depending on the timing and dose of the chemical agent. Copyright 2002 Wiley-Liss, Inc.
There are many others. My point is that there are clues, but there is not systematization.
In autism
Gene-environment interactions in mental disorders.
Br J Psychiatry Suppl. 2001 Apr;40:s12-7. Links
Understanding the roles of genome and envirome: methods in genetic epidemiology.Neiderhiser JM.
BACKGROUND: In order to understand studies of psychiatric epidemiology focusing on the 'genome' and 'envirome', basic knowledge of the logic and methods is necessary. AIMS: To provide a review of typical methods used in genetic epidemiology. METHOD: Reviews of the research designs usually employed in quantitative and molecular genetic studies. Genotype-environment correlation and interaction are also discussed. RESULTS: Quantitative genetic studies indicate that genetic influences are important for both psychiatric disorders and behavioural traits. Specific gene loci can be tested for associations with both psychiatric risk and behavioural traits by means of molecular genetic techniques. There has been little examination of genotype-environment correlation and interaction, although the few reports that have appeared suggest that these complex relationships are important. CONCLUSIONS: Advances in quantitative and molecular genetics now permit more careful examination of genotype-environment interaction and correlation. Studies combining molecular genetic strategies with measurement of the environment are still at an early stage, however, and their results must be awaited.
Genetic epidemiology (Still talking of hats)
A quotation
In conclusion, we have shown that failure to take an environmental factor into account may lead to erroneous conclusions about the relation between a genetic factor and a disease, when gene-environment interactions and competing risks are involved. The bias can sometimes be substantial, and we suggest some patterns of association in favor of this bias. These findings emphasize the need for application of rigorous epidemiologic principles in association studies. Researchers conducting studies on the etiology of late-onset diseases should be aware of this bias.
About role of epigenetics in autism and in general
Transcriptional control of cognitive development.
Folate and long-chain polyunsaturated fatty acids in psychiatric disease.
Human development:biological and genetic processes
Environmental and nutritional effects on the epigenetic regulation of genes.
Joseph, this is a very recent manuscript
Early blindness results in abnormal corticocortical and thalamocortical connections.
I deleted the same comment above because of problems with the links- even a space is enough to break the link (GRRR!).I think I have fixed them all.
Ma Luján
I found one link not working. Sorry
You can find the full text of the Eric Courchesne manuscript in a link of
Citation of pubmed of The developmental neurobiology of autism spectrum disorder
Hi Joseph,
The congenital blindness–autism connection is interesting.
From Hobson and Bishop 2003:
"one might consider that there are several sources of social impairment in blind children, including both physical and environmental factors, and that when potentiated by the children’s lack of vision, these result in specific forms of impoverishment in interpersonal experiences that have developmental consequences which include several autistic-like features."
One thing Dr Casanova did indicate in Casanova 2006 (as I interpret it – which is open to error), was that minicolumnopathy appears related to idiopathic autism, in suggesting it is not required when other identified medical conditions causing autistic-like symptoms exist – e.g. tuberous sclerosis, congenital rubella. Fragile X and Rett syndrome could probably be added to that list, as could at least some forms of congenital blindness. These conditions probably account for less than 10% of all cases of ASD. A better way of phrasing it is probably that "reduced minicolumn width appears to be a necessary prerequisite for idiopathic ASD and a factor driving neurodiversity." The former quote was my error, not one directly attributable to Dr Casanova.
Regarding blood flow, the links are interesting too, especially as they relate to 'structural' issues. As per my comment above, I would interpret these findings as ‘demand driven’, i.e. that ‘deficiencies’ or ‘abnormalities’ or ‘dysfunction’ (the authors’ words) resulted in less metabolic activity and therefore lower blood requirements rather than supply issues (i.e. deficiencies in blood supply) leading to brain damage and/or impaired function.
Regarding skull shape, is there a direct correlation to brain shape? Isn’t this phrenology? I believe that phrenology has been discredited. And is the issue brain shape, or developmental differences within various brain regions and the number, size and connectedness of the mincolumns and macrocolumns within those regions?
You wrote: "It’s clear that multiple factors are at play, which also makes it difficult to map genetically."
I totally agree. Linking minicolumn width as a prerequisite to idiopathic autism does not suggest that other factors are not at play. What I am instead suggesting is that that a brain with reduced minicolumn widths is vulnerable to ASD in a way that brains without reduced minicolumn widths are not, as evidenced by the fact that reduced minicolumn width is suggested as a prerequisite for autism but not sufficient by itself to cause autism. I would also mention the ‘T’ word as suggesting that genetics alone is not sufficient to explain autism (that genetic factors are a prerequisite is not in dispute here), but we’ve been there before (above) and I have sensory issues with hats.
You wrote: "The 'second hit' idea sounds like a vaccination event or something."
These are your words, not mine. I have not mentioned vaccines, nor have I mentioned timing (an event is presumably limited in time, place and scope), and I would suggest that it is inappropriate to try to link me at this point to either when I have not done so myself. ‘Second hit’ is a term I have seen in the literature referring to causes above and beyond the obvious genetic links, and where I’ve seen it the words were not used to specifically indicate or suggest a discrete event. Regarding "I think environmental influence works in more subtle and random ways.", I would not contest this.
Hi María Luján,
I think you missed a couple of papers. (grin)
"There are many others. My point is that there are clues, but there is not systematization.
In autism" Too true.
Wow, this is a wealth of information. I've been going through the various links, and I think my reading list is set for the next little while (says Ian as he bookmarks the comment for ease of access). This is a great list.
Thanks
"I wager "idiopathic ASD" is not a monolithic entity."
Agreed. I'm not saying anything different. I'm of the opinion that there are many different pathways to the same destination.
"Regarding skull shape, I don't actually mean phrenology."
(Grin) I know (says Ian, taking the opportunity to tease Joseph). Sometimes when the ball crosses the plate just so, you just have to swing at it. I agree with your last paragraph too, and would suggest that there is nothing said there that cannot easily and comfortably coexist with the minicolumn hypothesis.
Ian: let’s look for a moment at the requirements for a second hit to be material rather than inconsequential. One of the rationales for minicolumn existence is redundancy. So for a second hit to be material it must affect not just one or a small number of minicolumns, but must be significant in impact, and in effect must ‘permanently’ override normal development.
Okay, I'm back to the hats for a minute (admittedly). My point is that what constitutes a second hit is probably currently unknown (other than hypothesis) in most cases, and it's possible that this environmental influence could be a complex combination of factors. Invoking Occam's Razor (subjectively), if you introduce a billion variables during development, the simplest answer is that more than one variable plays a role. The more identifiable variables, the more chances for complex combinations that are actually simple occurences statistically. We simply don't know what constitutes significance in impact.
Ian, if you don't mind my asking, as you are obviously fresh on reading Casanova's work, how many brains have been imaged/examined in the course of his work. I'm honestly curious.
I'd like to read more of his work, if you're willing to pass along anything with his permission that won't make the copyright holders upset.
Hi Doc
Even when I agree with you in general, I also do think that there are very strong candidates that can affect the development, impacting the brain in a very negative way , even when the development can be affected with an enourmous amount of variables. For these candidates, there are a lot of published clues. And there is a lot of anecdotic evidence.
What is lacking is a systematized effort to understand the true impact of the strong candidates I mentioned, in ASD, with all the needed steps clearly visualized in a serious form with reproducible and trustable methods:detection, diagnosis and treatment.
At this point, again, I do not think the field is so nebulous.
María Luján
Fair enough Maria,
I'll need to dig into your list too, and do some reading. Thanks.
Hi Do'C
You said: "Okay, I'm back to the hats for a minute (admittedly)".
No, I don't consider that a hats comment. I would suggest that getting wrapped up in the MZ twins to the exclusion of the point of the post is hat related, but the existence of a ‘second hit’ is totally fair game given the implications that I have stated and my comment "e) that something else is therefore required (with no significant speculation by me yet as to what that something else may be, other than to generically label it as a ‘second hit’)". Regarding the 'billion variables', I’m much closer to María Luján’s "strong candidates". I plan to discuss this a bit more in follow-up post, soon to follow in Ian time (which, like dog years, can vary significantly from real time).
I’m not sure how many brains have been examined in total, but I do know that the work has been a) subject to peer review, and b) confirmed by others.
Regarding the rest, I’ll follow up via e-mail.
There's more to how the brain works than how it is structured, as well. Two people can use the same geographic part of the brain to do two different things. I suspect there are going to be substantial functional (not just structural) brain differences between autistic and non-autistic people. We don't have anywhere near the kind of scientific understanding yet to know exactly what those are. (We still can't 100% understand the data we get in functional imaging, we're not that advanced. There are those that claim to be, but... they're usually selling something.)
What I take issue with is the division between cognition and perception, or, as you put it, between "thinking" and "sensory issues". Perception is an important part of cognition for autistic people. It is not separate. There is no such thing as "sensory issues" that gets tacked onto "autistic cognition" like an afterthought causing the difficulties. It just doesn't work like that, it's not that tidy or simple. Any view that starts off with such a simplistic division doesn't tend to get much confidence from me that it's going to be right.
Hi Amanda,
Thanks for stopping by.
You wrote:
"There's more to how the brain works than how it is structured, as well. Two people can use the same geographic part of the brain to do two different things. I suspect there are going to be substantial functional (not just structural) brain differences between autistic and non-autistic people."
No dispute, depending on the level of structure that you’re talking about. At a minicolumn level the major structural difference appears to be narrower width (although again, still within the normal range). At a macro level, there are several documented structural variations within the brains of autistics vs. non-autistics, which are not in dispute here. Dr Casanova has discussed them in some of his work, but this post was specifically about minicolumns and autism, not about other brain variations. I would raise the possibility that variations in minicolumnar performance may drive some of these larger variations, as local connectivity variations would drive variations in global connectivity and subsequent brain development.
Regarding functional differences, it is definitely not beyond the realm of possibility. Given a greater degree of local vs. global processing in autistic brains, as well as neuroplasticity, plus the idea that "Similarities between minicolumns suggest that they are capable of performing similar transformations on incoming information, allowing them to replace each other in case of injury or to adapt to changing requirements" (quoted from this post), it definitely seems plausible that some functions may be performed with less connectivity than in non-autistic brains, and thus with functional differences within brain geography.
You wrote:
"What I take issue with is the division between cognition and perception, or, as you put it, between "thinking" and "sensory issues". Perception is an important part of cognition for autistic people. It is not separate. There is no such thing as "sensory issues" that gets tacked onto "autistic cognition" like an afterthought causing the difficulties. It just doesn't work like that, it's not that tidy or simple. Any view that starts off with such a simplistic division doesn't tend to get much confidence from me that it's going to be right."
Regarding the division between cognition and perception, the above statement has not been argued anywhere in this post (although I have argued it elsewhere). Dr Casanova’s research indicates that reduced minicolumnar width is a precondition for autism, and I layered on this that given that the reduced width is still within normal range and exists in those without autism (although they would presumably have variations in capabilities resulting from this structure - and to be clear, variations mean variations, not deficits), that this precondition is not sufficient by itself to make one autistic. If anything, Dr Casanova’s minicolumn argument does not differentiate between cognition and perception, but suggests that a deficit in inhibition may potentially affect both to a greater or lesser degree via a ‘short circuiting’ (my term) in minicolumnar functioning. I would suggest that minicolumnar inhibition deficits would be more inclined to result in sensory than cognition issues. But that point has not been argued here, so far, and definitely should not be attributed to Dr Casanova. On that point I do not know his thoughts.
Hi Ian,
Great post.
Sorry I am just now getting around to leaving a comment but I've linked to your post in my last blog entry.
WOW! That's a lot to take in, but it makes a great deal of sense. It also leaves some questions open, as does every theory. More research, though. Interesing to look at this, however, from a different perseptive - that of the inner workings of the brain.
Thanks, Ian
Just to let anyone know who shows up here via a link, there's a follow-on post on Autism and the Evolution of the Brain here.
Ian,
Doc asked if you had the info about how many brains have been imaged/examined, I am also interested in that answer.
Very interesting post.
Thanks
Post a Comment