In an earlier post I approached the subject of Autism, Genetics, and Evolution from the perspective of the requirements imposed by alleles and the theory of evolution. This post is an attempt to approach the subject from a different perspective, i.e. the evolution of the brain. My last post discussed autism and minicolumns, suggesting that a) ASD has a minicolumnar underpinning, b) this underpinning is required (i.e. no narrow minicolumns means no ASD), c) it originates in the first 40 days of fetal development (i.e. it is not itself acquired post-natally), d) that this difference falls within the normal range (i.e. that having it does not ‘cause’ a diagnosis, although it may very well result in diversity of thought and cognition, i.e. neurodiversity), e) that something else is therefore required (with no significant speculation as to what that something else may be, other than to generically label it as a ‘second hit’), and f) that research needs to prove or exclude causality among the population of the vulnerable, i.e. proving that something does not cause ASD in those who are invulnerable does not prove that it does not cause ASD in those who are vulnerable. This post builds upon the minicolumn post.
Dr Casanova also provided me with a copy of a work in progress on brain evolution and minicolumns (Casanova - Big Brains Manuscript, in preparation for submission), which has some interesting implications for autism, and prompted me to do some further reading. The same caveat applies as in my last post: I’m attempting to post about some of his work and add what I consider to be some of the implications. As before, bear in mind that I am not a neuroscientist, and as such there is the definite possibility that I have misinterpreted some of the ideas and findings. As such, any errors are mine. In addition, any implications beyond those explicitly stated in Dr Casanova’s research papers should not be attributed to Dr Casanova unless specifically noted.
Brain Size and Structure
A logical starting point in evaluating brain evolution is encephalization, or brain size. Statistical models have been built that compare body vs. brain size across species, from which one can derive an ‘expected’ brain mass based on body size. The actual brain mass divided by the expected brain mass yields an encephalization quotient (EQ). A result higher than one indicates a larger than expected brain mass, while a result less than one indicates a smaller than expected brain mass.
EQ is important as it allows for comparisons of brain sizes across species by automatically adjusting for body size. Larger animals would be expected to have larger brains, and elephants and some whales do have larger brains than humans. But after adjusting for body size, humans have much larger brains than other species. Using an EQ measure based on encephalization of insectivorous primates, humans have an EQ of 28.8, and there is a large gap between modern humans and all non-human primates, including the great apes (our closest relatives). This large gap is filled by the increasing encephalization of prehistoric hominid ancestors.
Encephalization has not affected all parts of the brain equally. The neocortex has been the primary beneficiary of this trend:
"The enlargement of the brain is not proportional; that is, all parts do not develop at the same rate. The neocortex is by far the most progressive structure and therefore used to evaluate evolutionary progress" (Stephan, 1972, p 174)
And Dr Casanova writes (Casanova - Big Brains Manuscript):
"It is the areal expansion of the isocortex and its connections that is ultimately responsible for primate encephalization. Ultimately, the simple addition of minicolumns is sufficient to explain cortical expansion, increased gyrification, and the subsequent parcellation and re-optimization of functional neural networks."
As brain size increases, connectivity requirements also change. Increases in cognitive capabilities may not result simply from increasing brain size or adding minicolumns, but rather, may be a function of brain connectivity. A key requirement for the brain is to generate a network of connections allowing for stable information processing while minimizing conduction and wiring costs. Connectivity is ‘expensive’ in terms of energy required to build and maintain connections. Bigger brains require both more connections and the interconnection of distant locations. Longer connections can result in signal delay/attenuation, and increase the probability of something going wrong. Increasing physical distance also raises the costs of connectivity, as the amount of energy required to maintain connections over longer distances is significant.
The answer to this connectivity issue appears to be at least threefold: modularization, gyrification, and parcellation.
By arranging neurons into minicolumns and driving connectivity between minicolumns, connectivity costs are reduced. Further organization of minicolumns into macrocolumns and then parcellation into functionally differentiated cortical regions also reduces overall connectivity requirements. The net effect is to allow a large number of cells to be connected by fewer axons and in more short-range connections.
Gyrification, i.e. the ‘folding’ of the cortex, reduces the physical distance between different areas and therefore the length of interconnecting fibers. Think of a piece of paper with two dots on it. The absolute distance between the two dots decreases as the paper is folded and the two dimensional surface in effect becomes three dimensional. Only one third of the human cortex is exposed to the surface, with the rest being found within sulci. Increased gyrification also increases the ratio of short range versus long rang connections within the brain.
Encephalization also results in an increase in parcellation of the brain into functionally differentiated cortical regions. Connectivity does not scale absolutely with brain size. As Dr Casanova writes: "increasing numbers of minicolumns impose a connectivity constraint, as non-adjacent or near-neighboring minicolumns become isolated by physical distance. Increasing distance limits connectivity as the amount of energy required to generate and maintain long-distance connections is substantial." Parcellation reduces the need for long distance connectivity by concentrating functions within locations. As encephalization increases, so does the amount of white matter dedicated to local connections. The result is a brain that is "not as equally densely connected relative to a smaller brain."
Within primates the corpus callosum, which connects the right and left hemispheres, generally becomes relatively smaller as isocortex size increases. As Dr Casanova writes, "Interestingly, at a gross anatomical level, perhaps the most obvious manifestation of increased parcellation is that the two cerebral hemispheres tend to be less densely interconnected and more independent." Connections between more distant regions are still required, and these may be maintained through specialized neurons, e.g. spindle neurons. As encephalization increases parcellation and decreases and/or channels connectivity into communication between larger units, there appears to be a corresponding increase in the number of spindle neurons to maintain functional communications.
The Costs of Encephalization
The human brain is expensive to build and maintain from an energy and nutritional standpoint. Kleiber’s law expresses the relationship between body mass and body metabolic requirements, i.e. resting metabolic energy requirements (RMR), also known as basal metabolic energy requirements (BMR). The equation is:
RMR = 70 * (W^0.75)
where RMR is measured in kcal/day, and weight (w) is measured in kg.
Mammalian brain size also scales with body mass, according to the formula:
E = 1.77 * (W^0.76)
where E is brain mass in grams. The similarity in the two exponential scaling coefficients is significant, implying that brain size and RMR are related, and a further inference is that the size of an individual’s brain is closely linked to the amount of energy available to sustain it (Foley and Lee, 1991).
While the brain is about 2.5% of our body weight, it accounts for 22% of our resting metabolism (Leonard and Robertson 1992, p 186). This is in sharp contrast to anthropoid primates using approximately 8% of resting metabolism for the brain, and other mammals using 3-4%. In contrast though, total human resting metabolism does not differ significantly from that of other mammals. So the question is, where does the metabolic energy to grow and sustain the human brain come from?
The answer is the ‘Expensive Tissue Hypothesis’, posited by Aiello and Wheeler, 1995. Aiello and Wheeler analyzed the ‘expensive’ (in terms of metabolic energy) organs in the body - the heart, kidneys, liver and GI tract – noting that together with the brain they accounted for the major share of total body BMR. Comparing the expected vs. actual size of these organs for an average 65 kg human, they found that there were significant differences in actual vs. expected size for both the brain and the gut.
They wrote (Aiello and Wheeler 1995, p 203-205):
"Although the human heart and kidneys are both close to the size expected for a 65-kg primate, the mass of the splanchnic [abdominal/gut] organs is approximately 900g less than expected. Almost all of this shortfall is due to a reduction in the gastrointestinal tract, the total mass of which is only about 60% of that expected for a similar-sized primate. Therefore, the increase in mass of the human brain appears to be balanced by an almost identical reduction in the size of the gastrointestinal tract…
"Consequently, the energetic saving attributable to the reduction in the gastrointestinal tract is approximately the same as the additional cost of the larger brain. "
Since the heart, kidneys and liver cannot be significantly reduced in size, due to their critical functions, to keep BMR at the expected level the higher energy costs of encephalization must be met by the gut. The implication for humans is that there had to have been an increase in dietary quality – e.g. more easily digested food, and the liberation of more energy/nutrients per unit of expended digestive energy - to allow for both a smaller gut and the reallocation of energy to encephalization.
Further evidence for the link between diet and brain size comes from the recent (in evolutionary terms) decrease in brain size in humans. Ruff, Trinkaus, and Holliday (1997) found that the human EQ reached its peak approximately 90,000 years ago, and has since remained fairly constant. But, absolute brain size has decreased by 11% since 35,000 years ago, with most of this decrease (8%) coming in the last 10,000 years. EQ has remained relatively constant because of an equivalent decrease in body size during the same timeframe.
So, what happened? The most plausible interpretation is that EQ is a genetically governed trait, and should not have changed materially in the last 10,000 years. This period has also seen the greatest social/cultural progress in human history, suggesting that the change is not the result of evolutionary selective pressure. Instead, the most likely change has been one of a shortfall in meeting nutritional requirements, in the form of one or more limiting factors preventing the body and brain from achieving their maximum potential development. The most obvious change in this period was the introduction of agriculture (i.e. the agricultural revolution), accompanied by a large rise in grain consumption and a significant drop in animal consumption (from perhaps 50% of diet to 10% in some cases).
The issue is presumably not caloric. Instead, the most plausible current hypothesis is that the decrease in animal consumption resulted in a consequent shortfall in consumption of preformed long-chain fatty acids (Eaton and Eaton 1998). For optimal growth the brain is dependent on the fatty acids DHA (docosahexaenoic acid), DTA (docosatetraenoic acid), and AA (arachidonic acid), which constitute over 94% of all HUFA (highly unsaturated fatty acids) in human and mammalian gray matter (Eaton et al 1998). Neuronal membranes are composed of a thin double-layer of fatty acid molecules. Loss in DHA concentrations in brain cell membranes correlates to a decline in structural and functional integrity of this tissue (source).
There is some evidence that humans are not able to synthesize sufficient levels of EPA (eicosapentaenoic acid – another omega-3 fatty acid) or DHA from precursor alpha-linolenic acid (LNA), and thus must get these EFAs from their diet. An impaired ability to synthesize EPA or DHA would increase the dietary dependence. All of the above are far more plentiful in animal foods than plants. Analysis of likely EFA intake during prehistoric times under a wide range of assumptions suggests that levels of EFA would have been sufficient (0.9g/1000kcal) to allow brain expansion and evolution (Eaton et al 1998). As the human diet changed, our access to these essential fatty acids declined.
Eaton et al 1998 discuss the differences in Paleolithic vs. current dietary EFA consumption, comparing a composite ‘diet of evolutionary adaptedness’ or DEA to current EFA intakes. Prior to the agricultural revolution, human consumption of DHA is estimated at 270 mg/day, while AA consumption is estimated at 1810 mg/day. Current estimates for Western diets are 80 mg/day and 100-1000 mg/day respectively, and the discrepancy for DTA is assumed to be similar. Paleolithic C18 PUFA consumption is estimated at nearly 21.5 g/day, with LA (linoleic acid) at 8.84 g/day and LNA (linolenic acid) at 12.61 g/day, with an overall omega 6:3 ratio of 0.70 in the C18 category. Current Western consumption is LA at 22.5 g/day and LNA at 1.2 g/day, with a total omega 6:3 ratio of 16.74. The result is a massive variation from EFA consumption during the period of peak human brain size, and may result in a shortfall in availability of key EFAs. Why does this shortfall matter?
As Cordain et al, 2001 explains, all mammalian brain tissue appears to have an invariant requirement for DHA and AA, without which normal neural function cannot occur: "Limitations to the supply of either one of these fatty acids will determine limitations to brain growth." They also indicate that the supply of these EFAs is constrained by the limited ability of the liver as well as other tissue to synthesize these fatty acids from their dietary precursors (LNA and LA). As such, we must consume the EFAs we need to build brain tissue. As the authors state: "Encephalization quotients decrease with increasing body size because there literally may be insufficient long chain fatty acid product (AA and DHA) to build more brain tissue."
To add a further note, while the researchers above have concentrated on the implications of EFA changes in our diets, it is also reasonable to assume that the changes in diet with the agricultural revolution would also impact access to other key nutrients, minerals, and even essential and conditionally essential amino acids, as discussed here. Access to zinc, iron, taurine, natural Vitamin A (as distinct from beta carotene) and B12 are some of the nutrients that would have been significantly impacted by the change in diet, although these changes have not been explored to nearly the same extent as the EFA issue. This is not to comment on the implications of these changes or to suggest any consequences or causality, other than to note that they exist and may have implications beyond those discussed in this post.
The increased cost of encephalization has also had another consequence for human development – delayed maturity. As part of their analysis, Foley and Lee, 1991 compare the development of human and chimpanzee brain growth patterns. Chimpanzees are born with a brain mass that is 47% of adult brain mass, and adult size is reached by four years of age. Humans, in contrast are born with brain masses that are 25% of adult brain mass, and by four years old have reached only 84.1% of adult mass. Despite a slower maturation, human daily brain energy costs start at twice those of chimpanzees, and by age five are 3.3 times higher. Growth costs are also commensurately higher.
Foley and Lee suggested that “Mothers from a variety of primate (and other mammalian species) have a goal of weaning infants at an optimal mass, ensuring those infants’ survival, and themselves producing again.” They theorize that the reason for delayed maturity in humans was an inability to sustain both higher brain energy costs and high rates of brain growth, leading to evolutionary selection for the energetically less demanding strategy of increasing the duration of human growth and maturity. Despite a higher DQ and increased foraging efficiency (through increased socialization and cooperation over time), human brain energy costs were sufficiently high to force a slowdown in the rate of growth, despite the cost of reducing female lifetime reproductive rates. Larger brains forced slower growth rates and delayed human maturation.
Brain Evolution and Autism
The evolutionary factors described above have some implications for autism. First, as I stated in the prior post, analysis has determined that minicolumns in the brains of autistic individuals tend to be smaller in size, although with the same total number of cells per column (Casanova et al, 2002a and Casanova et al 2002b). Given that autistic brains tend to be larger than average, the results indicate that autistics also have a higher number of minicolumns. In addition, the neurons within these individual minicolumns tend to be reduced in size. Reduced minicolumn width appears to be a prerequisite for autism. But, the reported minicolumn widths found within autistic brains are still within the normal distribution of minicolumnar width, albeit at the tail end (Casanova 2006). For lack of a better term, I’ll refer to this as a ‘pre-autistic brain’, i.e. meeting the structural requirements for idiopathic autism but not qualifying for an ASD diagnosis (perhaps the broader autism phenotype or BAP?). I’ll also assume that the pre-autistic brain neurons are also of reduced size, for reasons that I will discuss below.
Studies have also shown a postnatal acceleration in brain growth in autistics, resulting in increased brain volume in autistic children vs. controls (e.g. Redcay and Courchesne, 2005), Aylward et al 2002, among others). As per Hazlett et al, 2006, "Significant enlargement was detected in cerebral cortical volumes but not cerebellar volumes in individuals with autism. Enlargement was present in both white and gray matter, and it was generalized throughout the cerebral cortex. Head circumference appears normal at birth, with a significantly increased rate of HC growth appearing to begin around 12 months of age. CONCLUSIONS: Generalized enlargement of gray and white matter cerebral volumes, but not cerebellar volumes, are present at 2 years of age in autism. Indirect evidence suggests that this increased rate of brain growth in autism may have its onset postnatally in the latter part of the first year of life." Thus the autistic brain appears to have more minicolumns, more neurons generally, and appears to grow faster than NT brains.
Given that encephalization appears to result from the addition of more minicolumns, with a resulting increase in gyrification and parcellation, could a more densely packed autistic or pre-autistic brain be a continuation of this evolutionary trend? If so, then the adaptations related to encephalization would also be expected to increase, e.g. increased gyrification and altered parcellation. This appears to be the case. Hardan et al 2004 reported an increase in left frontal gyrification in autistic children and adolescents compared with controls. Herbert et al 2004 reported differences in the parcellation of white matter (Casanova - Big Brains Manuscript), as well as increases in cerebral white matter (Herbert et al, 2003). More minicolumns require more connectivity, requiring more white matter, increasing total brain size, and increasing the bias toward local over global connectivity. The smaller neuronal size within the autistic brain would further reinforce this local connectivity bias.
It may also be noteworthy that the larger than average brains of autistic individuals have consistently shown smaller corpus callosi. Basically, a 'pre-autistic' brain is potentially a more cell-dense brain that may have advantages but is less robust. The addition of more minicolumns could explain the larger brain, increased gyrification, and some variations in parcellation, all within the normal range (albeit at the tail end of the distribution curve), and all of the above would help explain an increased preference of this brain for local over global processing (continuing the encephalization-linked trend), even without any further impact that would tip this still non-autistic brain into ASD (hence the term 'pre-autistic').
I would hypothesize that this pre-autistic brain is another variation along the path of increased encephalization, created by genetic lottery to either survive and reproduce - if this 'model' ultimately has an absolute advantage or niche advantage - or wither if it does not. Could more minicolumns - but of narrower width - and more neurons but of reduced size be a 'normal' tradeoff related in part to metabolism and limits in connectivity and skull size? Larger neurons would have higher metabolic requirements, so smaller neurons might be a response to a resource constraint (i.e. they might have been larger if the resources to grow and sustain them were available). If the same number of minicolumns and neurons were of 'normal' size then this might also unduly strain connectivity and metabolic demands (connectivity over increasing distance results in increased metabolic requirements) and skull size past any reasonable limits (e.g. an increase in minicolumn width from 44.3 um to 67.8 um as per Figure 1 below would be a 53% increase). In this light, the pre-autistic brain can be viewed as an evolutionary attempt to increase the size of the brain within existing resource constraints.
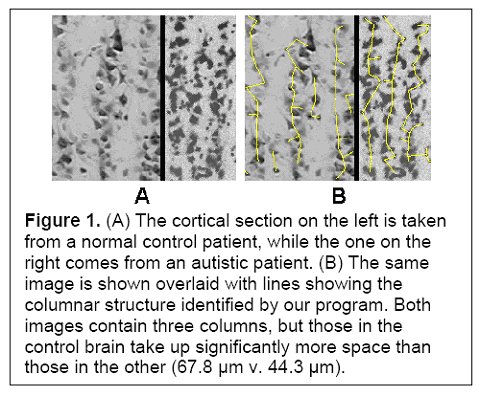
(Figure 1 Source – TMS Research proposal in preparation for submission, pg 2, with permission)
I would suggest that another reasonable hypothesis is that the pre-autistic brain could strain the nutritional capabilities of the body. If a normal brain constitutes 2.5% of body mass and accounts for 22% of BMR, (Wikipedia suggests that the developing infant brain consumes around 60% of the energy used by the body), then how much more would a larger and faster growing brain tax the metabolic system? Presumably a smaller neuron would require less energy or nutrition than a larger neuron, but I would suggest that the relationship is not linear, e.g. a neuron twice as large would require less than double the resources (in the case of the BMR, the exponential coefficient is 0.75, and it might be reasonable to expect something similar at a cellular level).
To the best of my knowledge it is an open question as to whether a pre-autistic brain also grows faster than average. A hypothesis is that the difference between the autistic and pre-autistic brain is the difference in growth rates. As stated in Courchesne, Redcay, and Kennedy 2004, “Head circumference, an accurate indicator of brain size in children, was reported to jump from normal or below normal size in the first postnatal months in autistic infants to the 84 th percentile by about 1 year of age; this abnormally accelerated growth was concluded by 2 years of age. Infants with extreme head (and therefore brain) growth fell into the severe end of the clinical spectrum and had more extreme neuroanatomical abnormalities.” Could increases in severity be a consequence of brain growth outstripping the required resources?
If autistic children have an increased rate of brain growth in the first few years of life then presumably this imposes a metabolic strain over and above the brain growth of NT children. Since no growth is ‘free’, this metabolic strain (energy and nutrients) must be somehow accounted for, either via an increased BMR, a reallocation of resources within the body, substitution (e.g. a deficiency in omega 3 EFA is compensated for by omega 6 EFAs, but at a functional cost), or through structural or operational deficiency. From Cordain et al, 2001 Fig. 2. “(Log-to-log chart of the resting metabolic rate of 20 anthropoids (new and old world monkey, apes and humans) relative to the predicted relationship based upon the Klieber equation)”, it is apparent that the actual to predicted BMR relationship is very close. I would suggest that BMR is a measure of efficiency that has been very tightly honed as a measure of evolutionary fitness (too low a BMR would result in energy constraints, while too high a BMR would increase the risk of starvation), and there is no reason to presume that autistics would vary significantly from non-autistics in adhering to expected results. But all of the other options result in potential downstream consequences in either the brain or other tissue.
If a shortfall in EFAs has potentially resulted in a decrease in size of the human brain by 11%, then what effect would the shortfall have on a pre-autistic brain with more minicolumns and an equivalent number of neurons per minicolumn – i.e. a brain with more neurons? Further, the increased rate of growth of the autistic (and pre-autistic?) brain in early childhood would increase even further the strain imposed by a shortage of EFAs. Smaller neurons, with a corresponding reduction in cell membranes and reduced metabolic demands might be a compensatory measure, gaining in metabolic efficiency and enhancing processing speed, the ability to process stimuli that require discrimination, and allowing for more complex information processing via local processing, but at a cost of reduced global processing and a reduction in ‘robustness’.
This tradeoff could still potentially have other consequences. Even with reduced neuronal size, the pre-autistic/autistic brain should presumably still require more energy and more nutrients than an NT brain, especially during the period of early childhood brain growth. If autistics adhere to the efficient tissue hypothesis then the logical tradeoff to satisfy this increased demand is with the GI system. And I would further speculate that another tradeoff may potentially occur with the liver.
Let’s be clear that I’m not suggesting at this point that the all autistic or pre-autistic children have GI or liver issues. What I am suggesting though is the following: a) that the increased brain costs must be met somehow, since no growth is ‘free’, b) that the GI system (and liver) are the logical organs to endure reductions to meet increased brain requirements, and c) that there in some cases may be consequences to this tradeoff in terms of the functional capabilities of these organs and potentially for overall health. In the case of the GI tract, some of these consequences could conceivably result in downstream issues (e.g. immune issues, impaired enzyme production, reduced metabolic efficiency, reduced nutrient and mineral absorption, etc.) that could have other systemic consequences, including consequences for the brain. Note that these issues would not ‘cause’ the brain to become ‘pre-autistic’, but they might have consequences for the pre-autistic brain that might result in an ASD diagnosis. Liver issues too could hypothetically have downstream consequences via a reduction in detox capabilities.
Further, if an NT brain is already facing resource constraints resulting in diminished size vs. potential (i.e. that 11% decrease in size), how much more critical would a GI 'issue' be for a pre-autistic brain? Even if the pre-autistic brain did not result in GI issues, comorbid GI issues might provide an additional resource constraint for a pre-autistic brain that could have a substantial impact on outcome. In this case it is not required that pre-autistic/autistic children have more GI issues than NT children, but rather, that given the increased metabolic demands of a pre-autistic/autistic brain, that the consequences of GI issues might be significantly greater on a larger and more demanding but less robust brain. The rates of comorbidity may be the same (although the efficient tissue hypothesis potentially suggests otherwise) but the consequences might not.
A further implication of the above goes back to the delayed maturity required of human offspring to allow for the management of the higher energy costs of encephalization. Since the autistic (and possibly the pre-autistic) brain is larger than an NT brain during the first few years of life and experiences a higher rate of growth, could delayed maturity be a further adaptive mechanism? Although much of this early growth is driven by connectivity, the increased number of neurons and minicolumns suggests that there is a lot more in the pre-autistic/autistic brain to connect, requiring substantial resources and energy. While this white matter growth is higher in autistics than NTs, it might still be a reduced rate of growth compared to what would be required to maintain an equivalent level of maturation in this larger brain as found in NT children.
Regardless of one’s beliefs regarding autism etiology, I would suggest that there are some serious implications regarding the nutritional status of autistics that flow from this analysis. The evolutionary and nutritional science referred to above is both mainstream and peer reviewed - no DAN! practitioners in sight. While the analysis has some similarities to Children With Starving Brains, the analysis, conclusions and hypotheses presented were arrived at from a different direction. I would suggest that it is a reasonable proposition that the autistic brain, if it is larger in childhood and goes through the growth spurt that mainstream research has detected, would have to face the same resource and energy constraints of NT brains, but with even higher requirements needing to be met. As such, one does not have to believe that addressing autistic nutritional requirements will result in a ‘cure’ to buy into the concept that autism brings with it some increased nutritional requirements, and that autistic brain ‘performance’ might be enhanced by compensatory nutritional intervention. Notice that I have ducked the entire issue of whether some autistic children with compromised diets due to sensory or tactile issues are further compounding any nutritional issues.
Finally, just to throw a hat into the discussion (see the last post’s comments regarding “The Meaning of Life” and hats for context), the above discussion may also offer a hypothesis to explain the ‘epidemic’, i.e. the perceived increase in the number of autistics in the past couple of decades. As mentioned above, absolute brain size has decreased by 11% since 35,000 years ago, with most of this decrease (8%) coming in the last 10,000 years. EQ has remained relatively constant because of an equivalent decrease in body size during the same timeframe. But in the post-WWII period human height has been increasing in the Western world and Asia. If EQ is genetically driven, then as stature increases, is brain size also on the rise? If resource constraints are lessening (perhaps with more variety in the diet, more access to protein, more access to EFAs?), then might constraints on pre-autistic brain size be diminishing along with constraints on human stature in general? Human stature can take at least two generations to recover (your grandmother’s dietary deficits affected your mother and the resources that she could bring to bear during your gestation). What if nutritional quality gains over the past two or more generations are now removing some of the nutritional constraints on pre-autistic brain development and size (i.e. more resources could allow for higher numbers of minicolumns to be generated during that first 40 days)? This might increase the pool of pre-autistic brains, i.e. the brains described above as being vulnerable to ASD.
Just something to think about.
